Predict FCD-lesion on MRI data
The new MELD pipeline offers a unique command line to predict a FCD-like abnormalities from T1w (and FLAIR scan).
Here is the video tutorial about how to run the MELD graph pipeline: Tutorial on how to run the MELD pipeline.
If you wish to use the harmonisation feature of the MELD pipeline, you will need to first have computed the harmonisation parameters for the scanner used to acquire the data and used the harmonisation code into the main pipeline command as described bellow. Please refer to our guidelines to harmonise a new scanner.
IMPORTANT new recommandation: We have received feedback regarding inconsistencies in MELD Graph results when using T1w+FLAIR scans compared to T1w scan alone. We advise users to primarily rely on T1w scans for lesion detection. If additional sensitivity is needed, FLAIR can be added to explore other potential clusters. However, these results will need to be interpreted with extra caution, as FLAIR-based clusters may include more false positives. For more information and guidance on how to run a second run with FLAIR see our FAQs.
Running
Ensure you have installed the MELD pipeline with docker container or native installation.
Ensure you have organised your data into MELD or BIDS format before running this pipeline
Ensure you have computed the harmonisation parameters if you want to use the harmonisation parameters
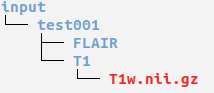
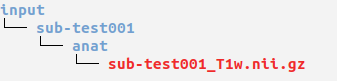
Open a terminal and cd to where you extracted the release zip.
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/new_pt_pipeline.py -id <subject_id>
WINDOWS USER: On windows, you do not need the DOCKER_USER="$(id -u):$(id -g)" part
Open a terminal and cd to the meld graph folder.
You will need to first activate FreeSurfer
export FREESURFER_HOME=<freesurfer_installation_directory>
source $FREESURFER_HOME/SetUpFreeSurfer.sh
Then run the command
./meldgraph.sh new_pt_pipeline.py -id <subject_id>
If using Singularity or Apptainer, there are some paths that you need to export before running the pipeline. Find the paths to export in the singularity installation. Tip : You can add those paths to your ~/.bashrc file to ensure they are always activated when opening a new terminal.
And then run:
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/new_pt_pipeline.py -id <subject_id>"
You only need to change the variable and path that are given in between “<>”. For example, change <meld_data> for the path to where your meld data folder is stored, and <freesurfer_license> for the path to where you have stored the license.txt from Freesurfer. See installation for more details
Tune the command
You can tune the MELD pipeline command using additional variables and flags as detailed bellow:
Mandatory variables |
Comment |
|---|---|
either |
if you want to run the pipeline on 1 single subject. |
or |
if you want to run the pipeline on more than 1 subject, you can pass the name of a text file containing the list of subjects. An example ‘subjects_list.txt’ is provided in the <meld_data_folder>. |
Optional variables |
|
|
provide the harmonisation code if you want to harmonise your data before prediction. This requires to have computed the harmonisation parameters beforehand. The harmonisation code should start with H, e.g. H1. |
|
use this flag to speed up the segmentation by running Freesurfer/FastSurfer on multiple subjects in parallel. |
|
use this flag to use FastSurfer instead of Freesurfer. (Requires FastSurfer installed for native installation). |
|
use this flag to skips the segmentation and features extraction (processes from script1). Usefull if you already have these outputs and you just want to run the preprocessing and the predictions (e.g: after harmonisation) |
More advanced variables |
|
|
use this flag to run to all the processes up saving the predictions as surface vectors in the hdf5 file. Does not produce produce nifti and pdf outputs. |
|
use this flag to do all the processes up to creating the prediction as a nifti file. Does not produce the pdf reports. |
|
use this flag to print additional information to debug the code (e.g print the commands, print errors) |
NOTES:
Outputs of the pipeline (prediction back into the native nifti MRI and MELD reports) are stored in the folder
output/predictions_reports/<subject_id>. See guidelines on how to interepret the results for more details.
Examples of use case:
To run the whole prediction pipeline on 1 subject using fastsurfer:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/new_pt_pipeline.py -id sub-001 --fastsurfer
./meldgraph.sh new_pt_pipeline.py -id sub-001 --fastsurfer
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/new_pt_pipeline.py -id sub-001 --fastsurfer"
To run the whole prediction pipeline on 1 subject using harmonisation code H1:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/new_pt_pipeline.py -id sub-001 -harmo_code H1
./meldgraph.sh new_pt_pipeline.py -id sub-001 -harmo_code H1
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/new_pt_pipeline.py -id sub-001 -harmo_code H1"
To run the whole prediction pipeline on multiples subjects with parallelisation:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/new_pt_pipeline.py -ids list_subjects.txt --parallelise
./meldgraph.sh new_pt_pipeline.py -ids list_subjects.txt --parallelise
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/new_pt_pipeline.py -ids list_subjects.txt --parallelise"
Additional information about the pipeline
The pipeline is split into 3 main scripts as illustrated below and detailed in the next sub-sections.
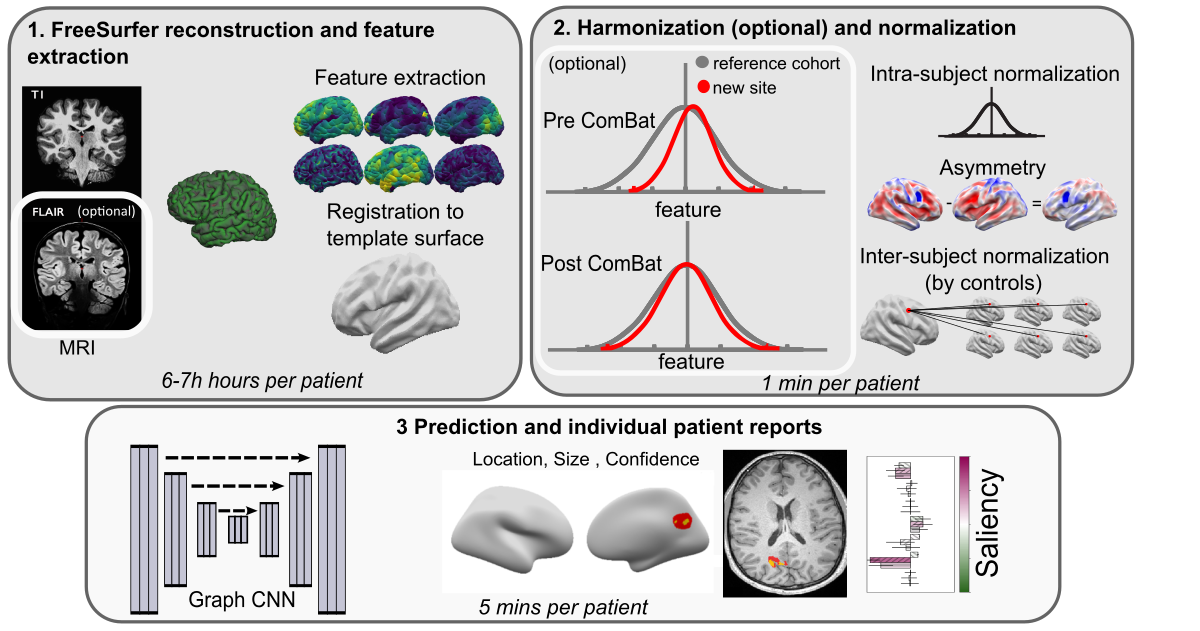
Script 1 - FreeSurfer reconstruction and smoothing
This script:
Runs a FreeSurfer/Fastsurfer reconstruction on a participant
Extracts surface-based features needed for the classifier:
Samples the features
Creates the registration to the template surface fsaverage_sym
Moves the features to the template surface
Write feature in hdf5
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_segmentation.py -id sub-001
./meldgraph.sh run_script_segmentation.py -id sub-001
First you will need to mount the meld_data folder to the /data folder of the container by running:
export APPTAINER_BINDPATH=<path_to_meld_data_folder>:/data
And then run:
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_segmentation.py -id sub-001"
To know more about the script and how to use it on its own:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_segmentation.py -h
./meldgraph.sh run_script_segmentation.py -h
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_segmentation.py -h"
Script 2 - Feature Preprocessing
This script :
Smooth features and write in hdf5
(optional) Combat harmonise features and write into hdf5
Normalise the smoothed features (intra-subject & inter-subject (by controls)) and write in hdf5
Normalise the raw combat features (intra-subject, asymmetry and then inter-subject (by controls)) and write in hdf5
Notes:
Features need to have been extracted using script 1.
(optional): this script can also be called to harmonise your data for new harmonisation code but will need to pass a file containing demographics information.
Example to use it on one patient without harmonisation:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_preprocessing.py -id sub-001
./meldgraph.sh run_script_preprocessing.py -id sub-001
First you will need to mount the meld_data folder to the /data folder of the container by running:
export APPTAINER_BINDPATH=<path_to_meld_data_folder>:/data
And then run:
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_preprocessing.py -id sub-001"
To know more about the script and how to use it on its own:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_preprocessing.py -h
./meldgraph.sh run_script_preprocessing.py -h
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_preprocessing.py -h"
Script 3 - Lesions prediction & MELD reports
This script :
Run the MELD classifier and predict lesion on new subject
Register the prediction back into the native nifti MRI. Results are stored in output/predictions_reports/<subjec_id>/predictions.
Create MELD reports with predicted lesion location on inflated brain, on native MRI and associated saliencies. Reports are stored in output/predictions_reports/<subjec_id>/predictions/reports.
Notes:
Features need to have been processed using script 2 and Freesurfer outputs need to be available for each subject
Example to use it on one patient without harmonisation:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_prediction.py -id sub-001
./meldgraph.sh run_script_prediction.py -id sub-001
First you will need to mount the meld_data folder to the /data folder of the container by running:
export APPTAINER_BINDPATH=<path_to_meld_data_folder>:/data
And then run:
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_prediction.py -id sub-001"
To know more about the script and how to use it on its own:
DOCKER_USER="$(id -u):$(id -g)" docker compose run meld_graph python scripts/new_patient_pipeline/run_script_prediction.py -h
./meldgraph.sh run_script_prediction.py -h
singularity exec meld_graph.sif /bin/bash -c "cd /app && source \$FREESURFER_HOME/FreeSurferEnv.sh && python scripts/new_patient_pipeline/run_script_prediction.py -h"
Interpretation of results
Refer to our guidelines for details on how to read and interprete the MELD pipeline results
FAQs
Please see our FAQ page for common questions about the pipeline use